원자는 끊임없이 움직이고 있지만, DFT는 모든 것을 0 K에서 정지시킨다. "자발적인" 반응, 생성, 또는 합성을 정확하게 기술하려면 내부 에너지가 아닌 자유 에너지가 필요하다.

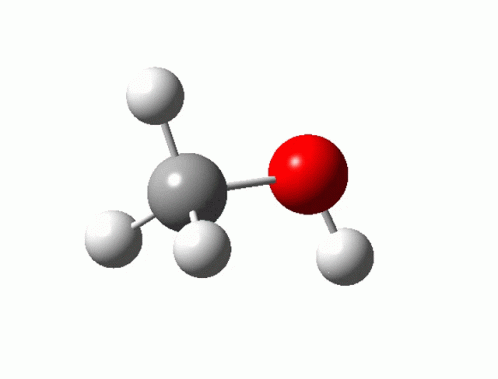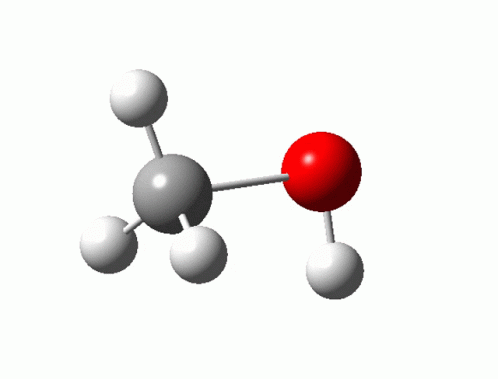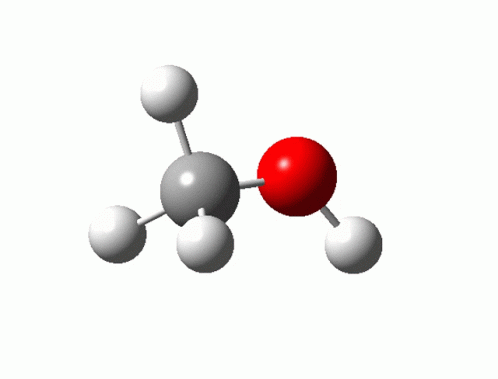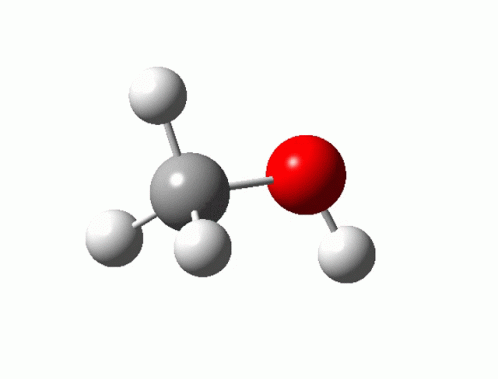
1$E_{\text{DFT}}$정적 0 K, ZPE 없음
2$E_0$$+ \text{ZPE} = \sum \tfrac{1}{2}h\nu_i$
3$G(T, P)$$+ H_{\text{thermal}} - TS$ (진동, 배치)
4$G_{\text{ec}}(U)$$- neU$ (CHE) + 전기장 효과, 용매화
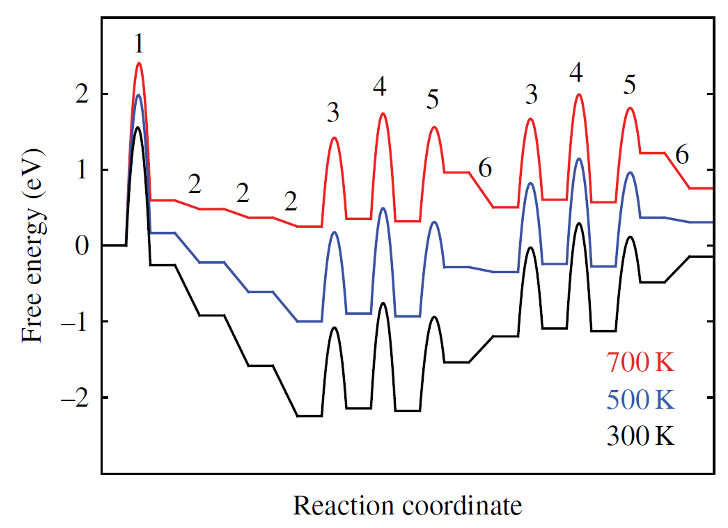
NH3 합성 — 부호 반전 (700 K)
ΔH0 = −0.95 eV
ΔG0 = +0.49 eV
Pt 위의 CO — 엔트로피가 지배
ΔE = −1.5 eV
ΔG ~ −0.8 eV at 500 K
- 평형 피복률 θi(T, P)
- 에너지 스팬 모델을 통한 TOF
- 선택성: 생성물은 ΔE가 아닌 ΔG에 의해 결정됨
- TST: $k = \frac{k_B T}{h} e^{-\Delta G_{\text{TS}}/k_B T}$
ΔG는 화학의 올바른 통화이다. EDFT는 진공 상태의 0 K 근처에서만 유효하다.
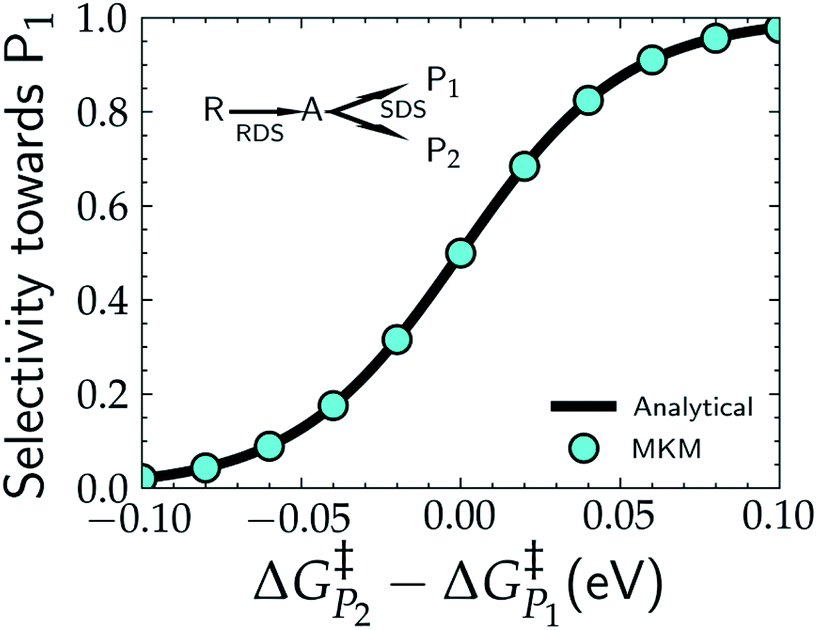
선택성은 $\Delta G_{P_2}^{\ddagger} - \Delta G_{P_1}^{\ddagger}$의 $\pm 0.05$ eV 범위 내에서 급격하게 전환된다
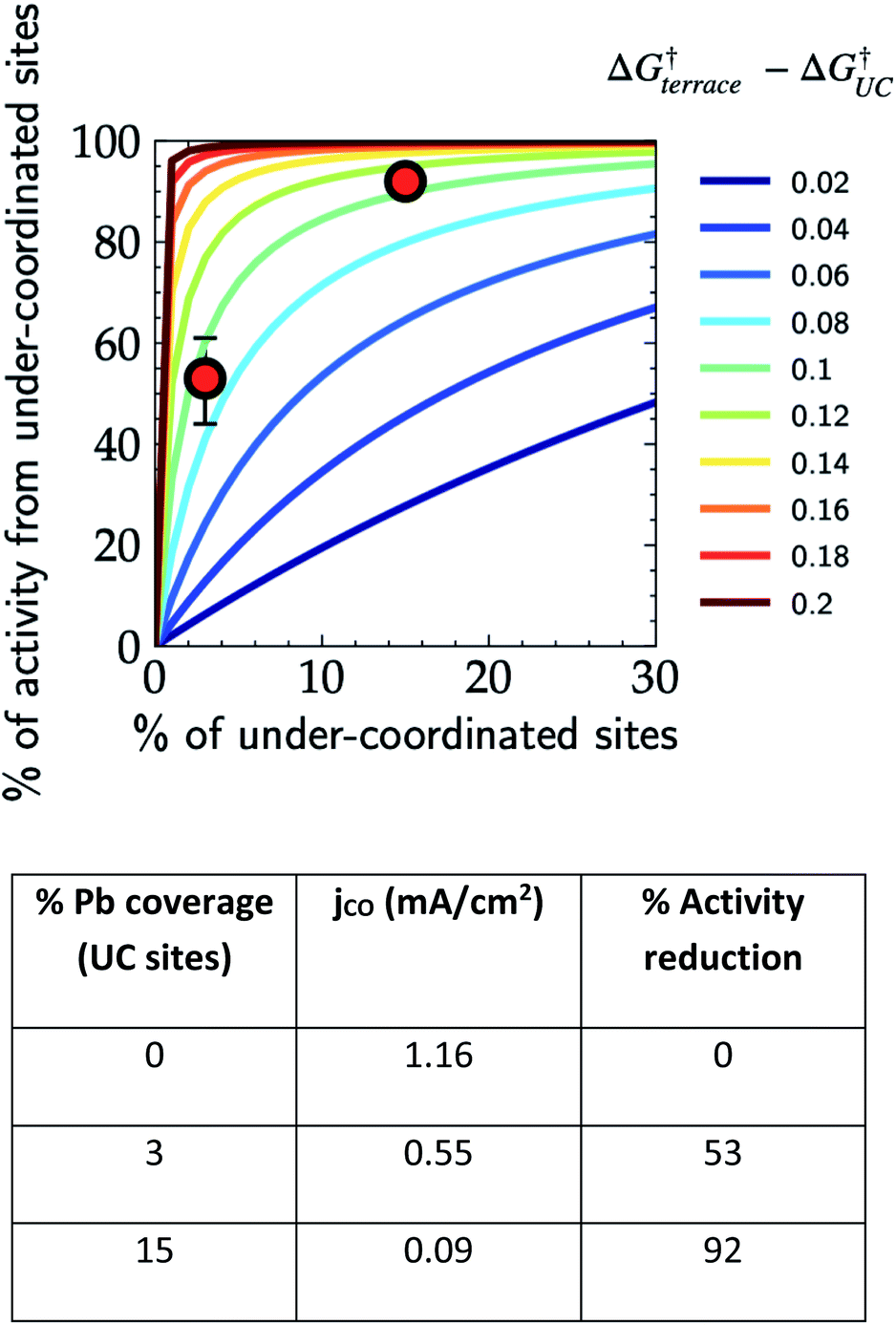
저배위 사이트의 전체 활성에 대한 기여도 대 풍부도 및 $\Delta G_{\text{terrace}}^{\dagger} - \Delta G_{\text{UC}}^{\dagger}$. 빨간 마커: Au(111)에서 Pb-UPD를 이용한 CO2RR, −0.6 V vs. RHE.
$$\text{TOF} = A \cdot e^{-\Delta G^{\ddagger}/RT}$$
일반적인 DFT 오차 ~0.15 eV는 이미 TOF에서 ~300배 오차로 이어진다.
작은 자유 에너지 차이 (0.05–0.10 eV) — 종종 DFT 오차 범위보다 작음 — 가 어떤 생성물이 형성되고 어떤 사이트가 지배하는지를 결정한다. $G$를 올바르게 구하는 것은 선택이 아니다.
그림 출처: Govindarajan, Kastlunger, Heenen & Chan, Chem. Sci. 2022, 13, 14–26. DOI: 10.1039/D1SC04775B
$E_{\min} = \tfrac{1}{2}h\nu$는 퍼텐셜 우물 바닥 위에 있다 — Heisenberg 불확정성 원리는 정지한 입자를 허용하지 않는다
ZPE 공식
- $\text{ZPE} = \sum \tfrac{1}{2}h\nu_i$, 모든 실수 진동 모드에 대해 합산
- N–H, O–H (3000 cm⁻¹ 이상): 0.2–0.5 eV 기여
- 저주파수 제한된 모드 (200 cm⁻¹ 미만): 무시 가능
- 상태 간 ZPE 변화만 중요 (ΔZPE)
정량적 주의사항
- NH3 합성: ZPE 없이 0.83 eV 과도하게 발열 (Norskov 표 2.1)
- H 이동 반응: 항상 ZPE를 포함할 것
- 무시해도 되는 경우: H 이동이 없는 무거운 흡착종 (ΔZPE 0.05 eV 미만)
CO2 → CO (2e⁻)
U = −0.10 VΔG = +0.20 eV/e⁻
TΔS ≈ 0 — 양쪽의 기체 몰수가 동일; 엔트로피가 도움을 주지 않음
CO2 → CH4 (8e⁻)
U = +0.17 VΔG = −0.17 eV/e⁻, 자발적
큰 유리한 ΔH가 엔트로피 벌칙을 압도; 전자당 열역학적으로 가장 용이
엔트로피 없는 DFT는 유한한 T와 P에서 CO2RR 생성물 선택성을 잘못 순위 매길 것이다
상 다이어그램은 T, P, pH, 또는 U의 함수로서 어떤 상태가 가장 낮은 G를 갖는지를 나타낸다. 안정성 경계: $\Delta G_{AB} = 0$
구성 방법
- 각 경쟁 상에 대해 $G(\text{variable})$를 작성
- 각 조건에서 가장 낮은 $G$ → 안정한 상
- 교차점이 안정성 경계를 정의
- 변수: $T, P(\text{O}_2)$; $U, \text{pH}$; $\mu_C, \mu_N$
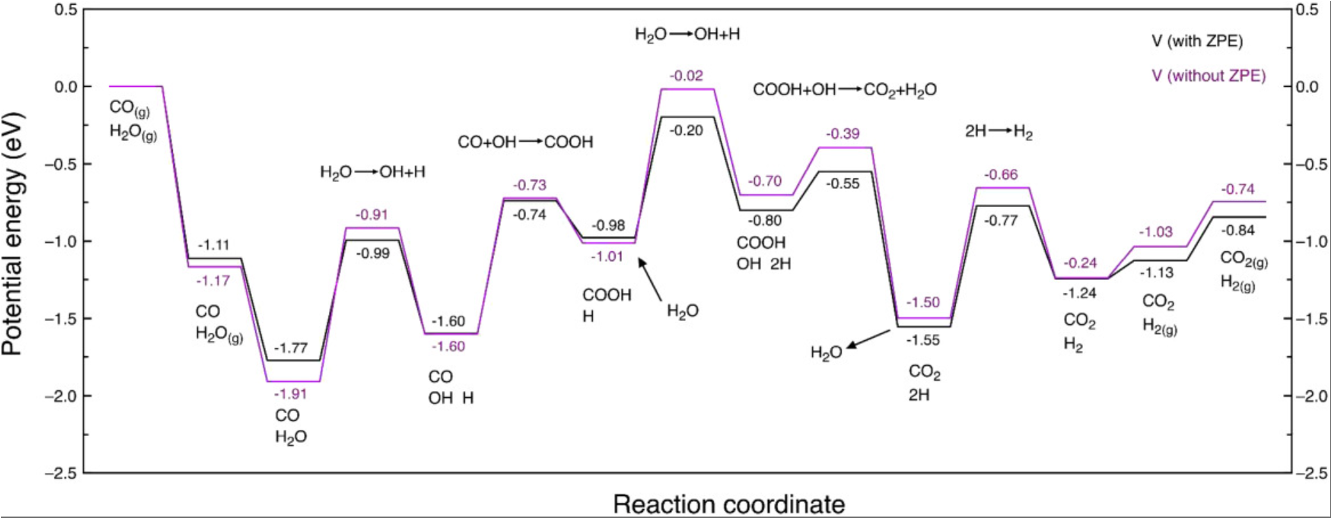
예시: PdOx/CeO2 CH4 산화
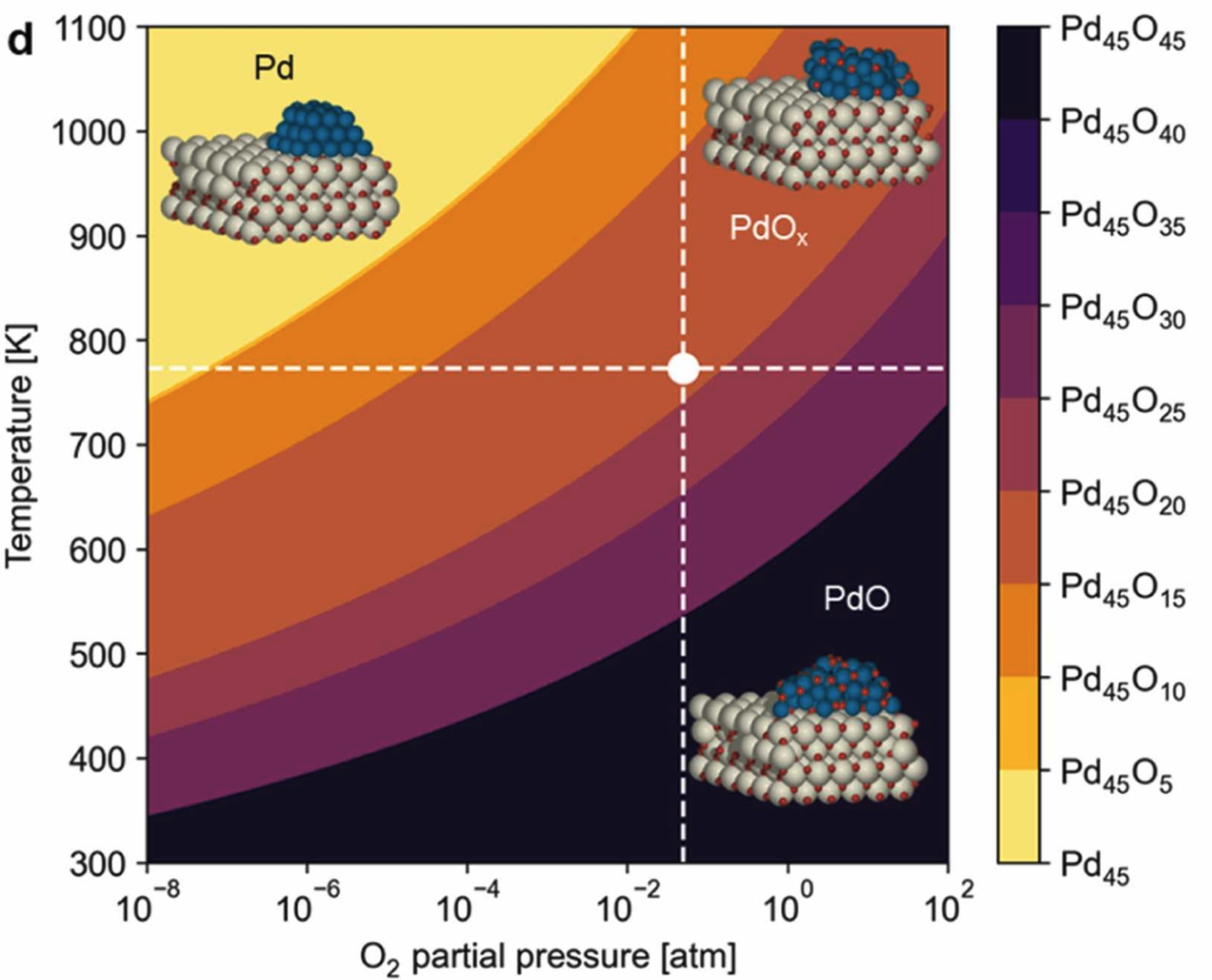
Ryu, Choung et al. Appl. Catal. B 379 (2025) 125672
$\Delta G_{\text{ox}}(T, P_{\text{O}_2}) = \Delta G_f^{\circ}(\text{PdO}) + \tfrac{1}{2}RT\ln(P_{\text{O}_2}/P_{\text{ref}})$
- 결함이 풍부한 CeO2는 Ce3+ 전자 공여를 통해 부분 환원된 PdOx를 안정화 — 경계를 더 높은 T로 이동
- 상 경계에서 활성 상태; 동적 산화환원 펄스가 일시적으로 이를 가로지름
$$G(U, \text{pH}) = G_{\text{DFT}} + \text{ZPE} - TS - neU + n \cdot k_B T \ln(10) \cdot \text{pH}$$
구성 방법
- 각 표면 상태에 대해 $G(U, \text{pH})$를 작성. 기울기는 $n$ (이동된 전자 수)에 의해 결정.
- 각 $U$에서 가장 낮은 선이 안정함. 교차점 → 상 경계.
- 2D 맵을 위해 pH 0–14를 스윕. pH는 298 K에서 0.0592 V/pH만큼 이동.
예시: Fe(OH)x/Pt ORR
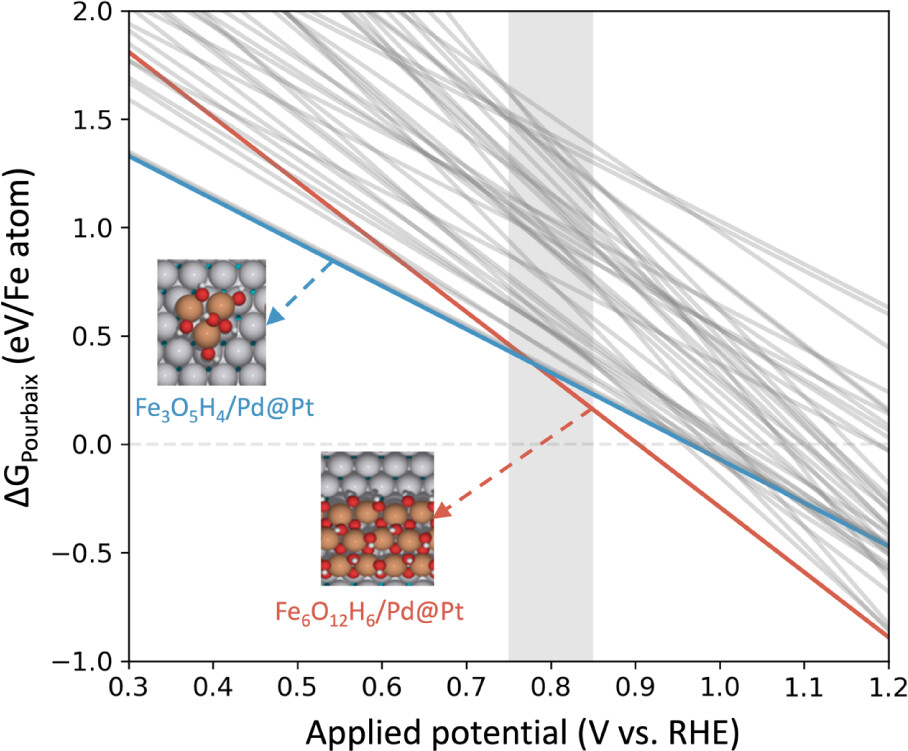
Maiti, Choung et al. ACS Appl. Mater. Interfaces 17 (2025) 40517
- ORR 조건 (0.7–0.9 V vs. RHE)에서: Fe3+(OH)3 또는 FeOOH가 안정 — 금속 Fe가 아님
- Pt 기판이 전하 이동을 통해 벌크 Fe Pourbaix 대비 경계를 이동시킴
- 작동 전위에서 Fe3+ 산화 상태를 고정하도록 계면을 설계
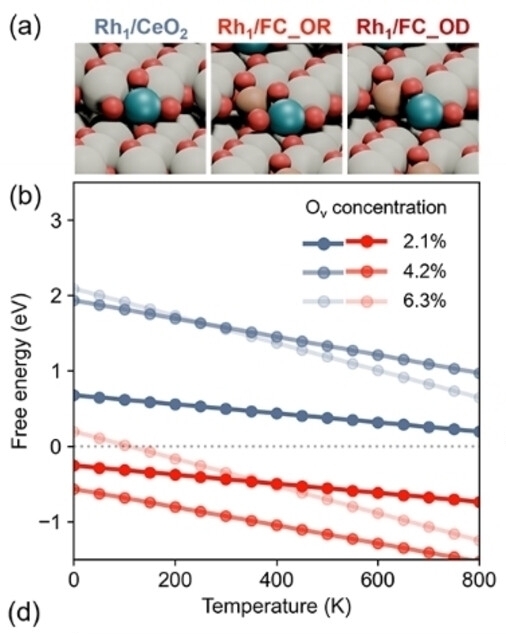
Kim, Choung et al. Angew. Chem. Int. Ed. 64 (2025) e202421218
Rh+ SAC가 반응 조건에서 Rh⁰로의 소결 대비 안정하게 유지되는가?
경쟁 상태: Rh+/결함, Rh3+/화학량론적, Rh⁰ 클러스터, 용해된 Rhn+
- Ce3+ 결함은 Rh+를 화학량론적 사이트보다 ~0.5–1.0 eV 더 강하게 결합
- 결함이 풍부한 Fe-Ce 산화물은 높은 T에서 더 낮은 P(O2)까지 Rh+ SAC 안정성 영역을 확장
- 상 다이어그램이 소결 저항성을 직접 예측 — 내구성 실험으로 확인됨
- 결함이 풍부한 지지체에서만 작동점이 SAC 안정 영역 내에 있음
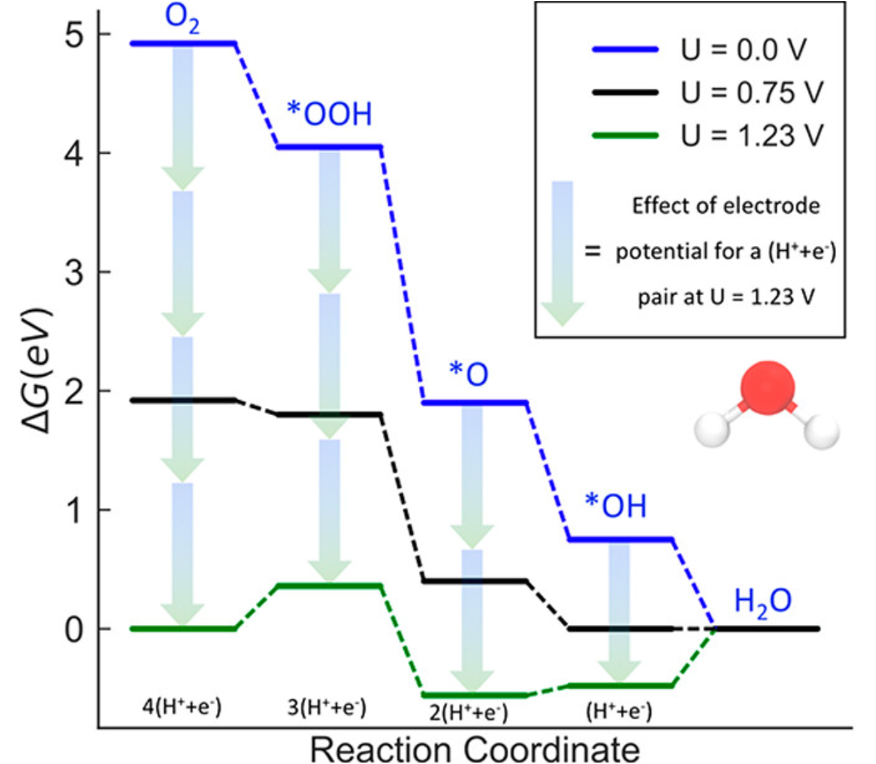
1O2* → OOH*ΔG ~ +0.4 eV (U = 0 V) — Pt에서의 전위 결정 단계
2OOH* → O* + H2OΔG ~ −0.2 eV (발열)
3O* → OH*ΔG ~ −0.5 eV (U = 0 V)
4OH* → H2OΔG ~ −0.8 eV (U = 0 V); U = 0.80 V에서 흡열 — Pt에서 ~0.35 V 과전위를 설명
제한 전위 UL은 마지막 흡열 단계가 0에 도달하는 지점이다. 과전위: η = Ueq − UL = 1.23 − UL
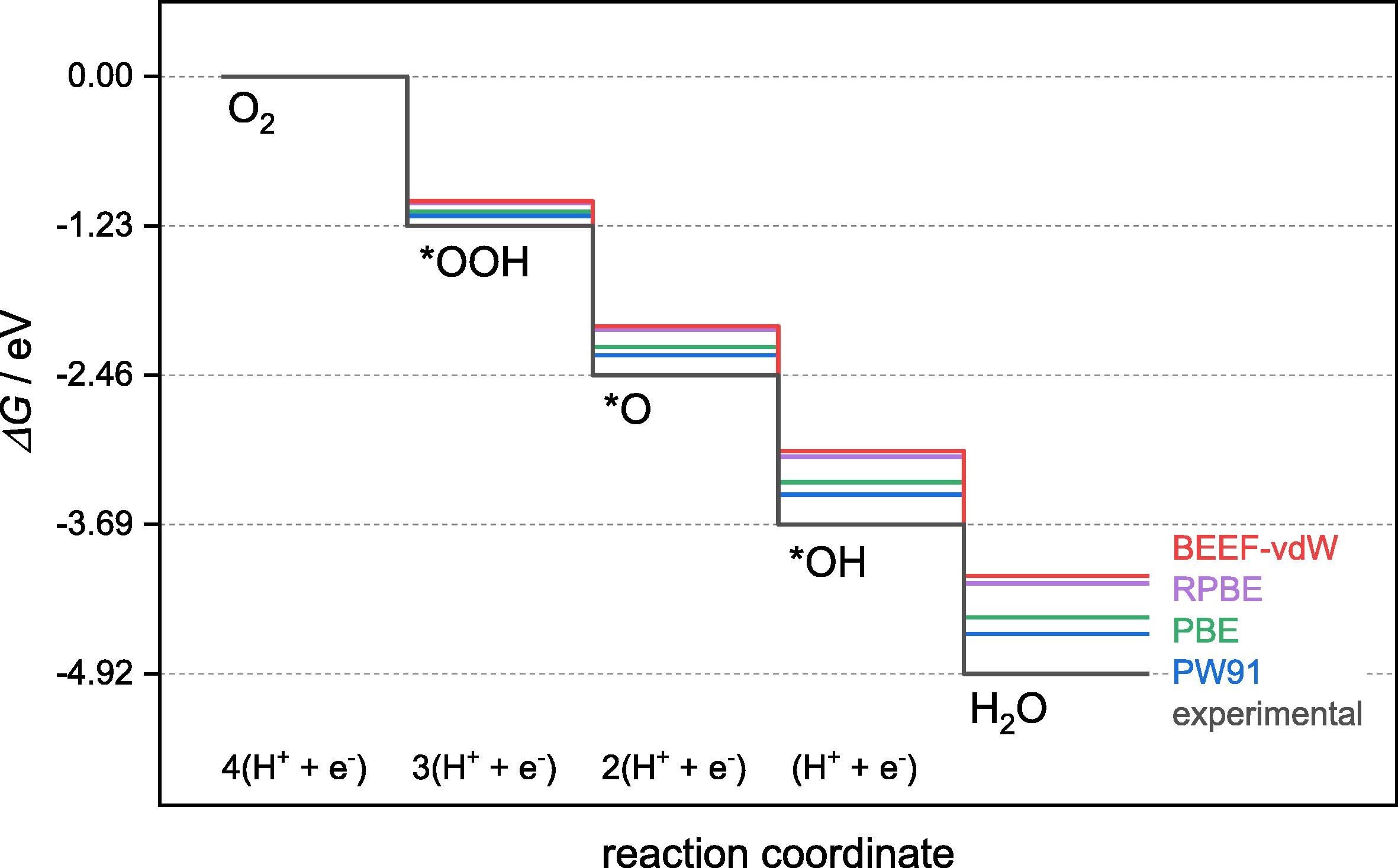
Sargeant, Illas, Rodríguez & Calle-Vallejo, J. Electroanal. Chem. 896 (2021) 115178
GGA-PBE는 O 함유 흡착종을 과결합함
금속 위 O*, OH*, OOH*에 대한 체계적 오차 0.2-0.4 eV. 무작위가 아님 — 유사한 흡착종 간에 상관되어 있어 화산 플롯은 여전히 촉매를 올바르게 순위 매김. 절대 과전위는 0.2-0.4 V 오차가 있을 수 있음.
주의할 때
- 실험 대비 절대 흡착 에너지
- 서로 다른 작용기 비교 (O vs. N 흡착종)
- 산화물 — GGA+U 또는 HSE06 필요
오차가 상쇄될 때
- 같은 흡착종 계열 내의 상대 에너지
- 화산 플롯 순위 (오차가 상관됨)
- 스케일링 관계 기울기 (범함수에 거의 무관)
Govindarajan, Kastlunger, Heenen & Chan, Chem. Sci. 2022, 13, 14–26
- DFT 스크리닝은 일반적으로 하나의 결합 사이트를 선택한다. 선택한 사이트가 동역학적으로 지배적인 사이트가 아니면, 수치적 정확도와 무관하게 예측이 실패한다.
- Au(111) Pb-UPD 등고선 분석 (Nitopi et al., Chem. Sci. 2022): 테라스와 저배위 사이트 간 80-120 meV 장벽 차이가 Arrhenius를 통해 저배위 사이트에서 20-100배 높은 TOF로 변환됨.
- Co 산화물 OER: 소수 결함 사이트가 평균 Co 표면 사이트보다 100배 높은 TOF를 보임 (Frei 시간 분해 FTIR; Plaisance 미시 동역학 모델링). 지배적인 활성 사이트는 인가 전위에 따라 변화함.
- 0.10 eV 장벽 차이는 일반적인 DFT 오차 범위보다 작지만, 속도를 제어하는 사이트를 1-2 자릿수 변화시킨다. 사이트 정량 (Pb-UPD, 산화환원 피크 적분, CO/CN 프로브)이 필수적이다.
- 완벽한 DFT 동역학조차 실험에서 물질 전달 제한(H2, O2, CO2의 낮은 용해도)에 의해 가려질 수 있다.
CHE는 문헌 어디에나 있다 — 무엇을 할 수 없는가?
- 동역학적 장벽 없음 — CHE는 순수 열역학적; 실제 장벽은 0.3-0.8 eV 다를 수 있음
- 명시적 용매 없음 — 묵시적 용매화는 극성 중간체에 대해 ΔG를 0.1-0.3 eV 이동시킬 수 있음
- 피복률 의존성 없음 — 측면 상호작용이 개시 전위를 0.1-0.2 V 이동시킴
- Frumkin 보정 없음 — 이상적인 이중층을 가정; 높은 과전위에서 실패
- Grotthuss 호핑을 포함하는 양성자 이동 단계에 부적합 — 명시적 물 분자 필요
CHE는 순위 매기기와 스크리닝에 탁월 (대략 +/- 0.30 V 정확도). 정량적 실험 비교를 위해서는 미시 동역학 모델링과 명시적 용매화를 병행할 것.