Research
Raising AI scientists for materials, experiments, and processes. Accelerating chemical engineering from atoms to industry.
We develop chemical engineering AI platforms that connect atomistic foundation models, scalable simulation, generative models, AI agents, and experimental data. We integrate graph neural networks, atomistic simulation, generative models, scientific agents, and experimental feedback into a unified platform for materials discovery and process optimization, from hypothesis to deployment.
1 AI for Catalyst Discovery
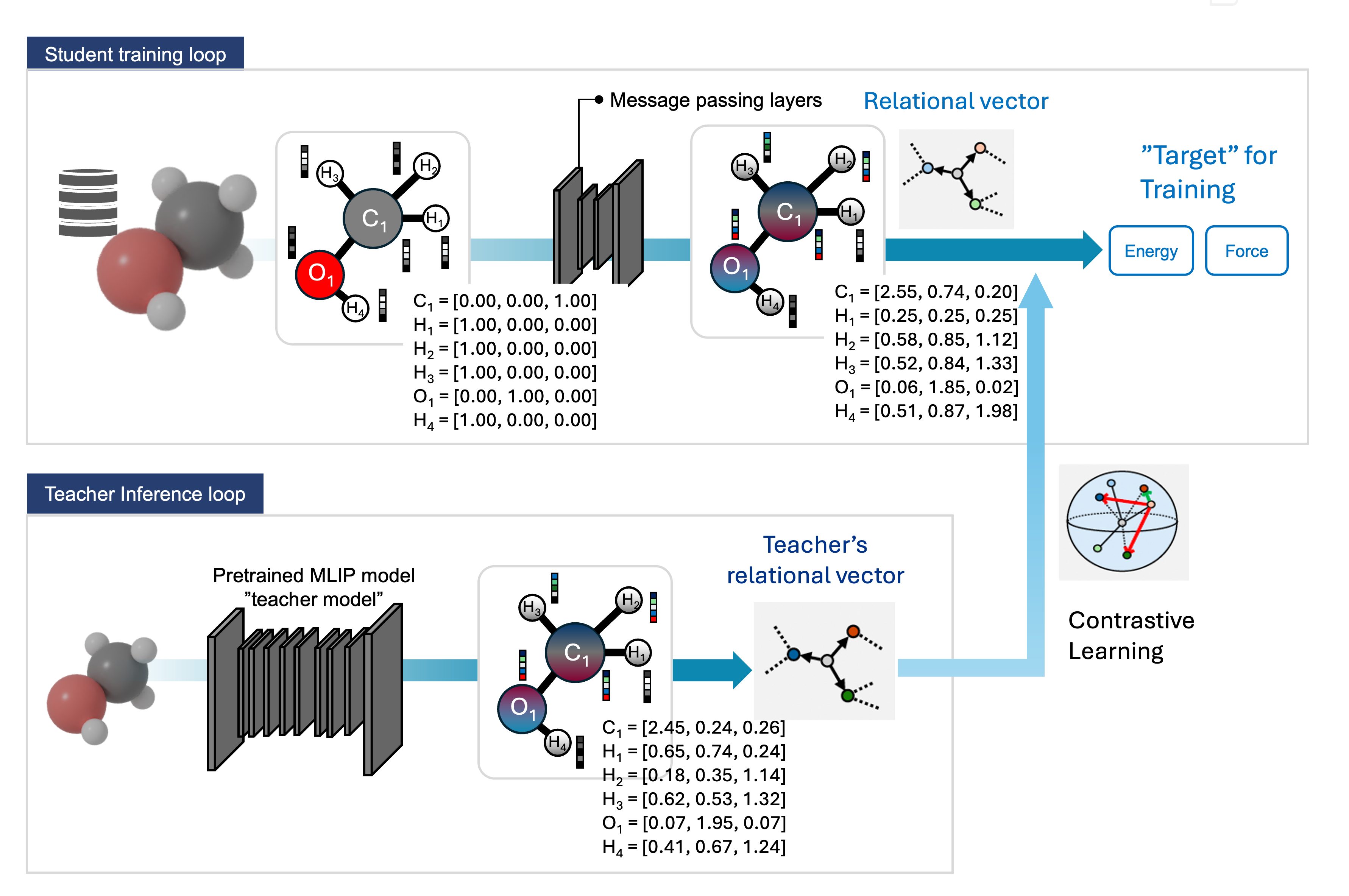
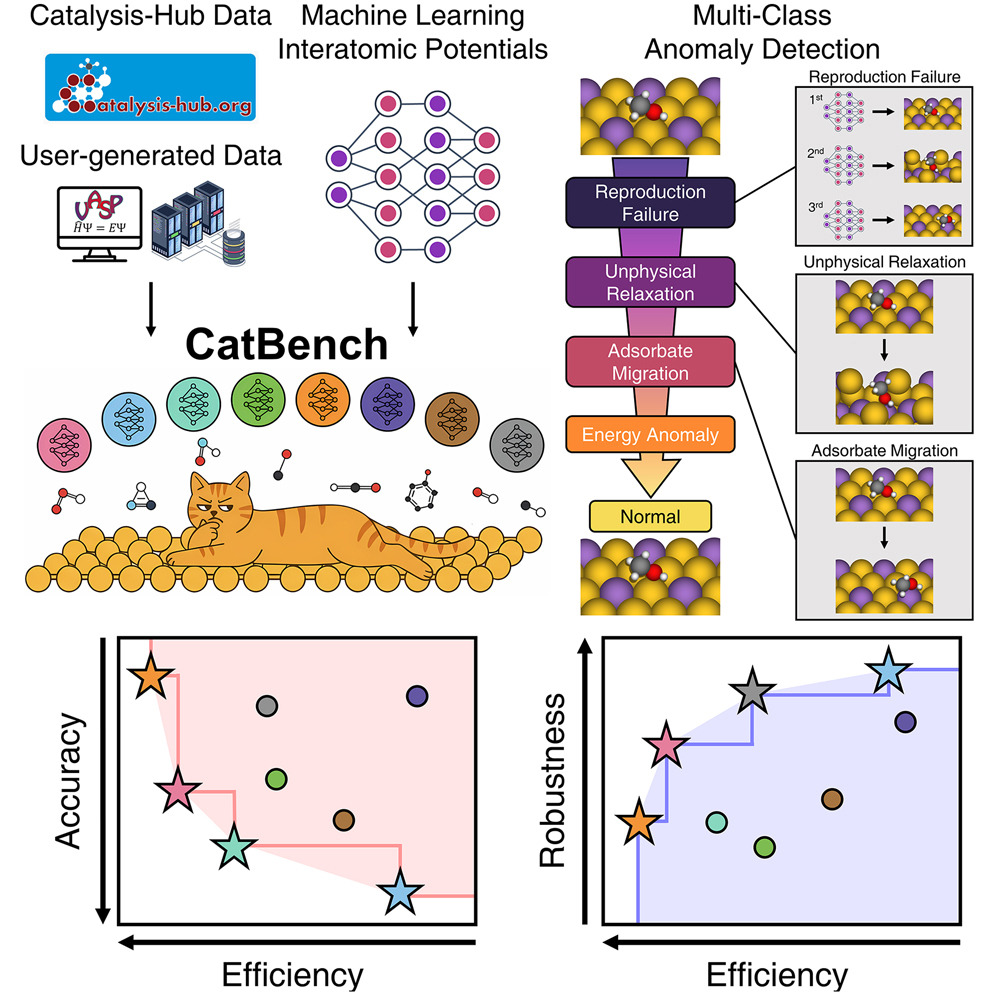
Developing practical machine learning tools for accelerating catalyst screening at scale. ARK (Angular Relational Knowledge Distillation) compresses large teacher MLIPs into 7× smaller student models with 50× speedup, preserving angular interaction fidelity critical for surface reactions. CatBench systematically evaluates 14 universal MLIPs on 47,000+ adsorption reactions with multi-class anomaly detection, revealing failure modes invisible to standard metrics.
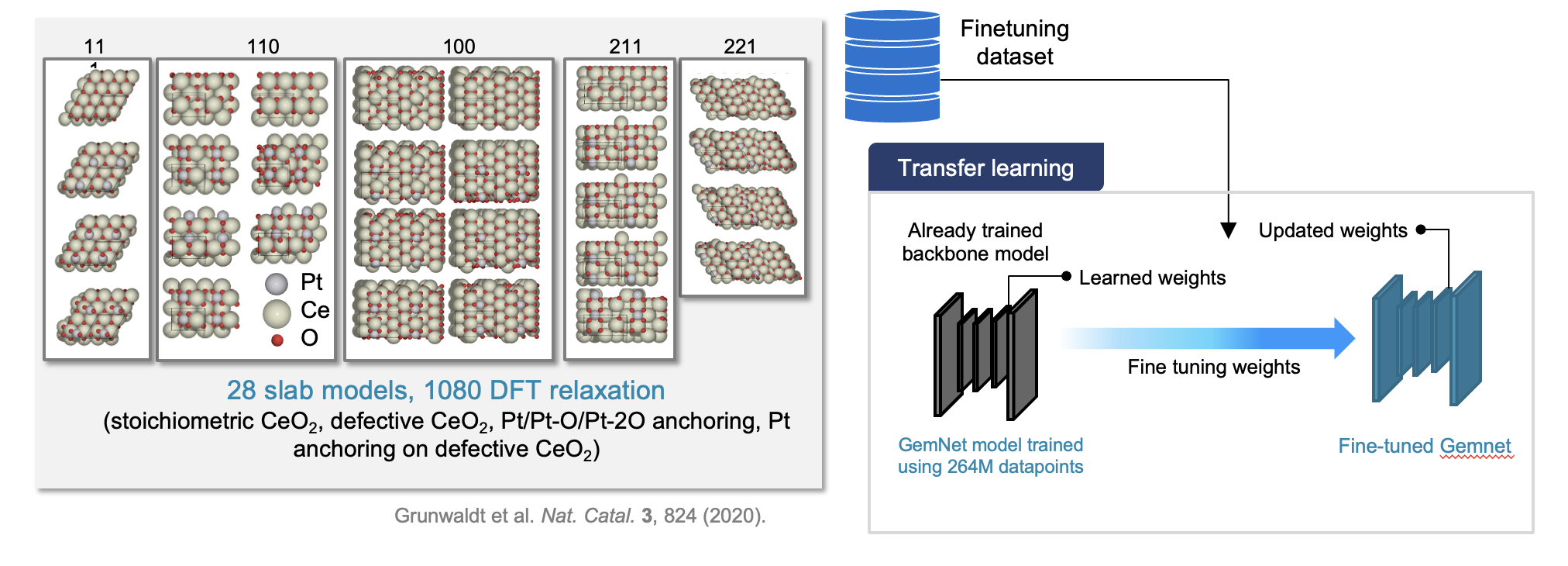
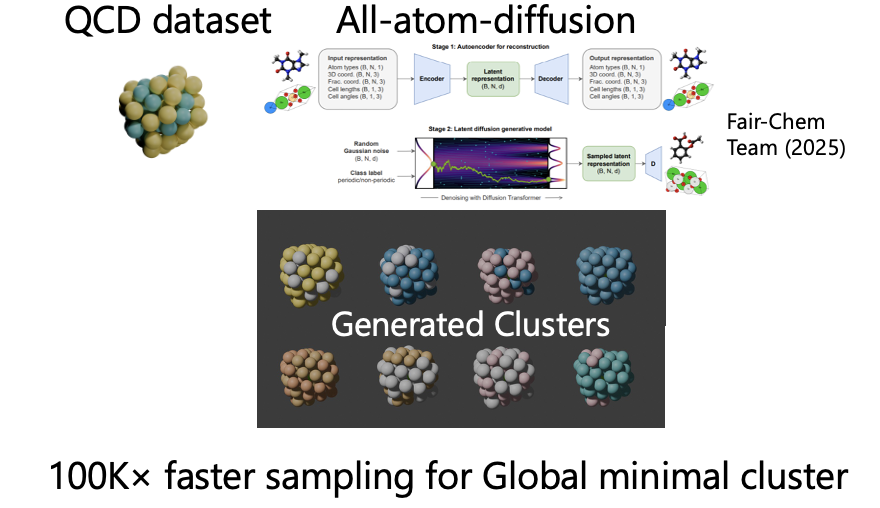
We also develop domain-specific finetuning pipelines that adapt pre-trained MLIPs to target catalytic systems achieving DFT-level accuracy from limited training data, and generative diffusion models for designing thermodynamically stable bimetallic nanocatalysts. CatAgent, our autonomous multi-agent LLM framework, orchestrates hypothesis generation, surrogate-model evaluation, and critic feedback for electrocatalyst discovery, achieving 2.27× discovery rate over random search.
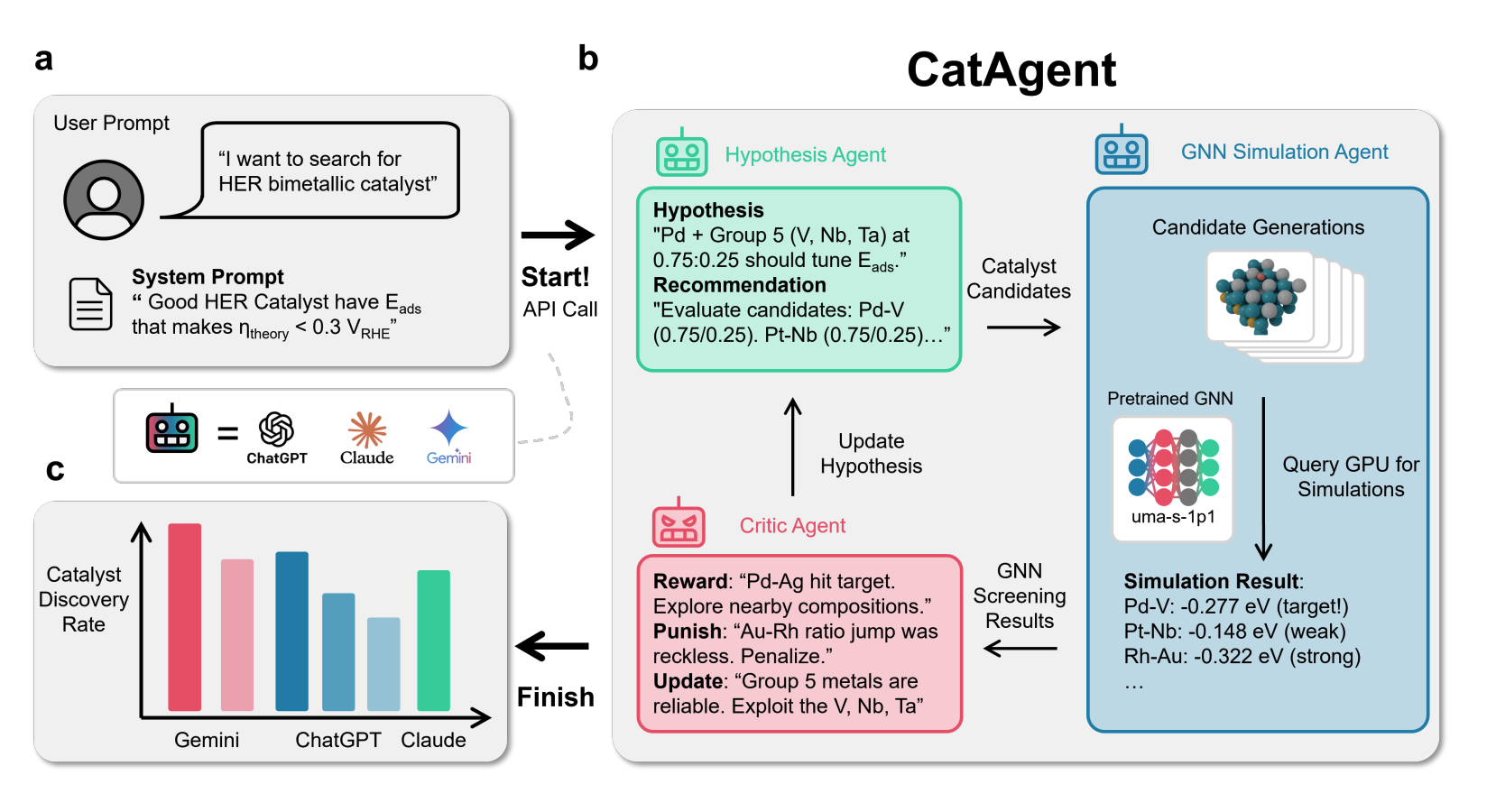
Selected Publications
-
Angular relational knowledge distillation of machine learning interatomic potentials for scalable catalyst exploration
npj Computational Materials, Accepted (2026) · Earlier version accepted at AI4Mat-NeurIPS 2025 -
CatBench: Benchmark framework of MLIPs for adsorption energy predictions in heterogeneous catalysis
Cell Reports Physical Science, 6, 102968 (2025) -
CatAgent: Multi-agent orchestration for electrocatalyst discovery
ICLR 2026 Workshop AI4Mat, Accepted (2026) -
Rise of machine learning potentials in heterogeneous catalysis: Developments, applications, and prospects
Chemical Engineering Journal, 494, 152797 (2024)
2 Operando Simulation & Experiment
Closing the loop between AI predictions and experimental reality. Using MLIP-driven molecular dynamics, we simulate complete catalyst nanostructures up to 10,000 atoms at nanosecond timescales — bridging atomic-scale DFT accuracy with mesoscale dynamics that traditional surface slab models cannot capture.
Our multiscale approach spans from single-atom catalysts on oxide supports to realistic nanoparticle morphologies, revealing size-dependent oxygen transfer mechanisms, lattice oxygen kinetics in hierarchical ceria architectures, and facet-dependent reconstruction under reaction conditions. We are extending this framework to electrochemical interfaces, modeling explicit solvation, proton-coupled electron transfer, and electrolyte ion effects at catalytic surfaces. All predictions are validated by ambient-pressure XPS, in-situ Raman, XANES, and synchrotron characterization through direct collaboration with experimentalists.
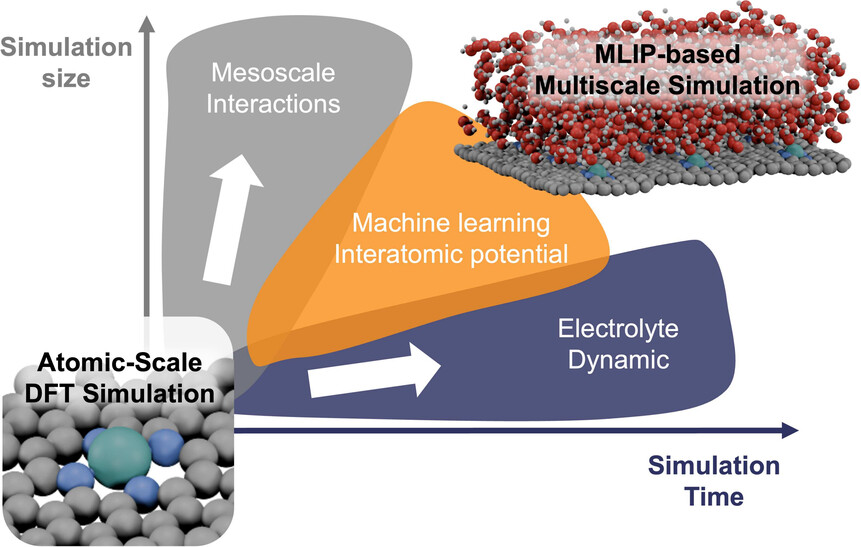
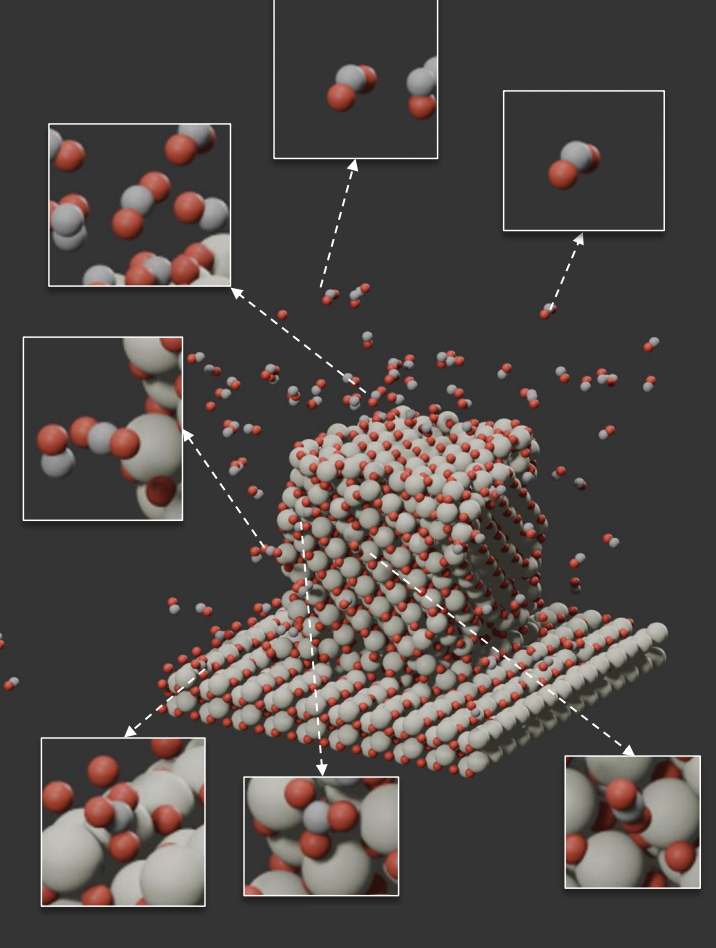
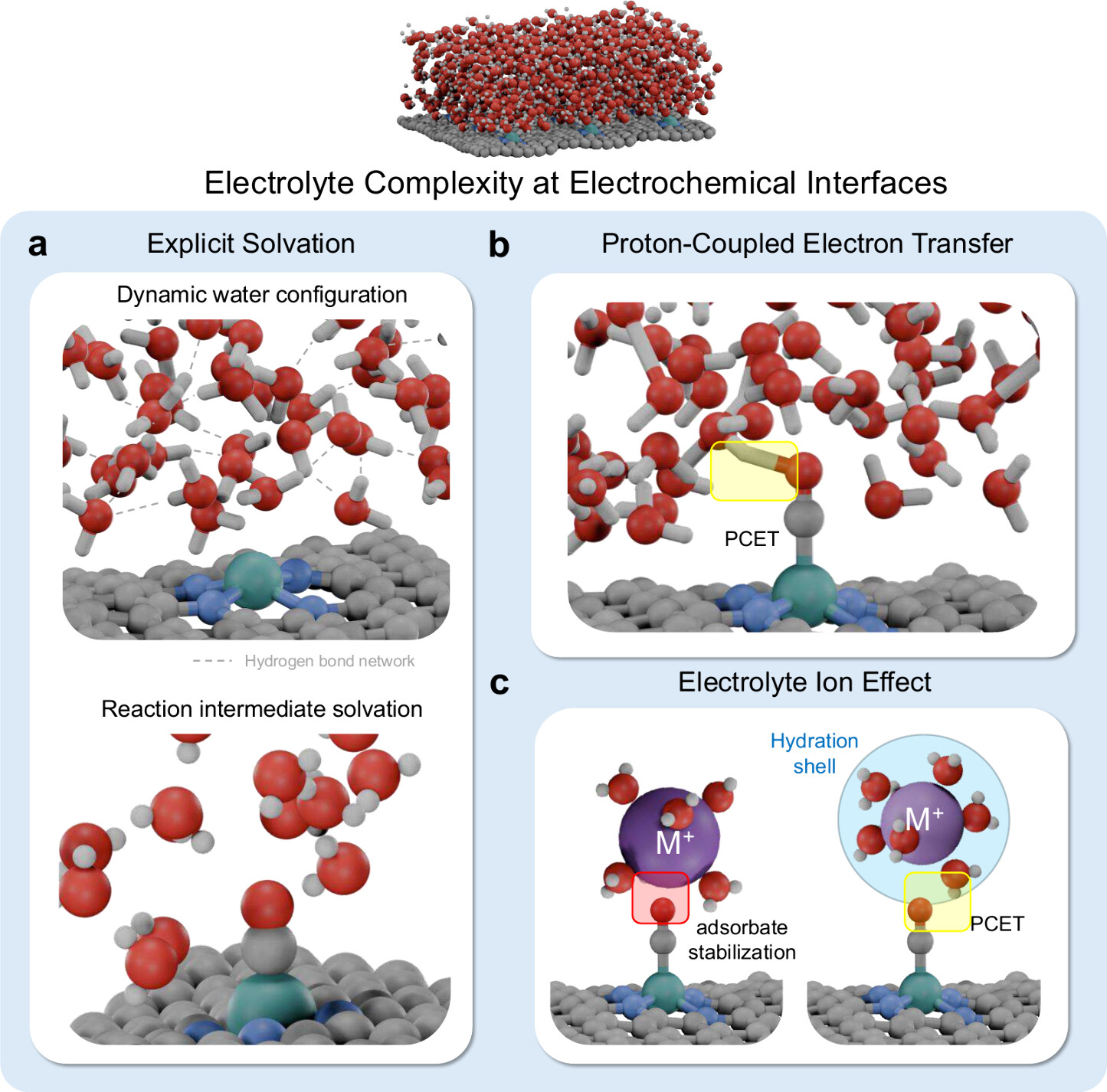
Selected Publications
-
Understanding oxygen transfer on ceria with Pt single atoms for surface reaction
Nature Communications, 17, 298 (2026) -
Hierarchical ceria nanoarchitecture enabling accelerated lattice oxygen activation for efficient redox reactions
Nature Communications, 17, 5868 (2026) -
From atomic motif to realistic single atom catalysts through machine learning interatomic potentials
ACS Energy Letters, 10, 6288-6296 (2025) -
Partially reduced PdOx nanoparticles strongly interacting with defect-rich ceria via dynamic redox pulse for complete methane oxidation
Applied Catalysis B: Environmental, 379, 125672 (2025)